Compare two lineage definition matrices
plot_lineage_defs2.RdCompare two lineage definition matrices
Usage
plot_lineage_defs2(
left_def,
right_def,
col = c("#b2182b", "#f7f7f7", "#2166ac", "grey90", "white", "grey40"),
...
)Arguments
- left_def, right_def
Lineage definition matrices, such as those produced by
astronomize()andusher_barcodes().Alternatively the output ofprovoc(), from which the lineage definition matrix will be extracted (one may be a lineage definition matrix and the other may be the result of fitting provoc, allowing for comparison of which lineages/mutations were actually used).- col
A vector of three colours (left, middle, right). For a mutation present in
left_defonly, the colour will be the first colour in the vector, while a mutation not present will be 80% of the way between the left colour and the middle colour (similar forright_def). Optionally, the user can specify a vector of length 6, where the colours define left, middle, right, NA, neither, and both.- ...
Arguments passed on to
heatmap(), such asmainfor a title andmarto give the title space.
Details
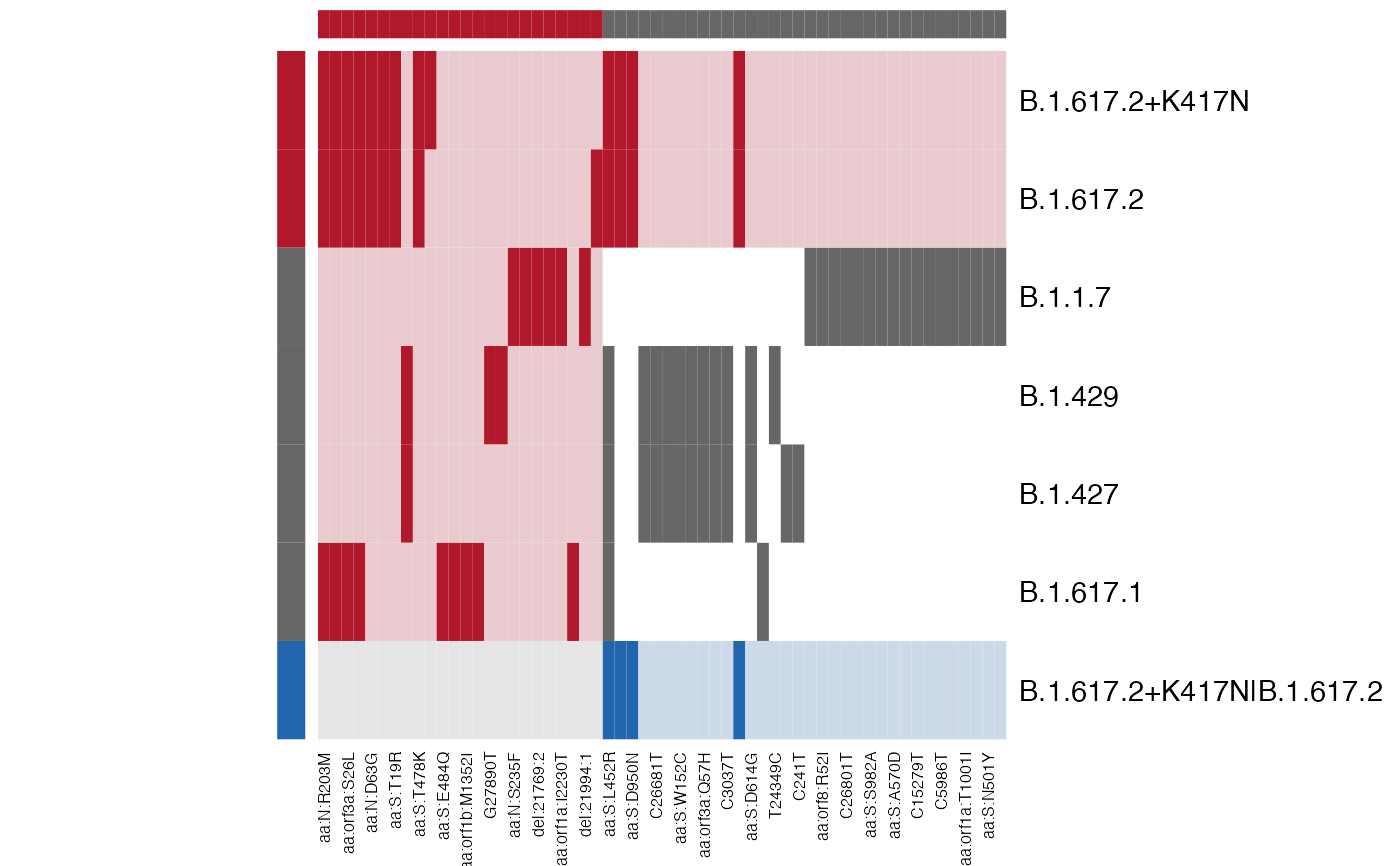
A darker colour means that the mutation is present in that lineage, whereas a lighter colour means it's absent.
The first colour in col represents mutation/lineage combinations that are only present in the left definitions, and the third color in col means it's only in the right.
The center square represents mutations and lineages that are present in both lineage definitions, with white indicating that the mutation is not present for that particular lineage and black indicating that it is present.
Examples
lineages <- c("B.1.1.7", "P.1", "B.1.617.1", "B.1.617.2", "B.1.617.3")
left_def <- astronomize() |> filter_lineages(lineages[-1])
right_def <- usher_barcodes() |> filter_lineages(lineages[-2])
#> usher_barcodes.csv not found. Downloading from Freyja repository.
#> Writing to ./usher_barcodes.csv
plot_lineage_defs2(left_def, right_def,
main = "Constellations (without B.1.1.7) vs. Usher Barcodes (without P.1)")
#> Error in total_def[lins_lmr$mid, muts_lmr$right] <- right_def[lins_lmr$mid, muts_lmr$right] + 4: incorrect number of subscripts on matrix
plot_lineage_defs2(left_def, right_def,
main = "Constellations (without B.1.1.7) vs. Usher Barcodes (without P.1)",
col = c(2, 0, 3))
#> Error in total_def[lins_lmr$mid, muts_lmr$right] <- right_def[lins_lmr$mid, muts_lmr$right] + 4: incorrect number of subscripts on matrix
# See which mutations/lineages were actually used
lineage_defs <- astronomize() |>
filter_lineages(c("B.1.617.1", "B.1.617.2", "B.1.617.2+K417N",
"B.1.427", "B.1.429", "B.1.1.7"))
res <- provoc(count / coverage ~ ., data = b1,
lineage_defs = lineage_defs)
# B.1.617.2 and B.1.617.2+K417N were "squashed" because they're identical.
# We can also see which mutations were *not* used in the analysis.
# They are the ones defined in the left_def but not present in right_def.
plot_lineage_defs2(lineage_defs, res)